









Mikrokeemia
Mis see on?
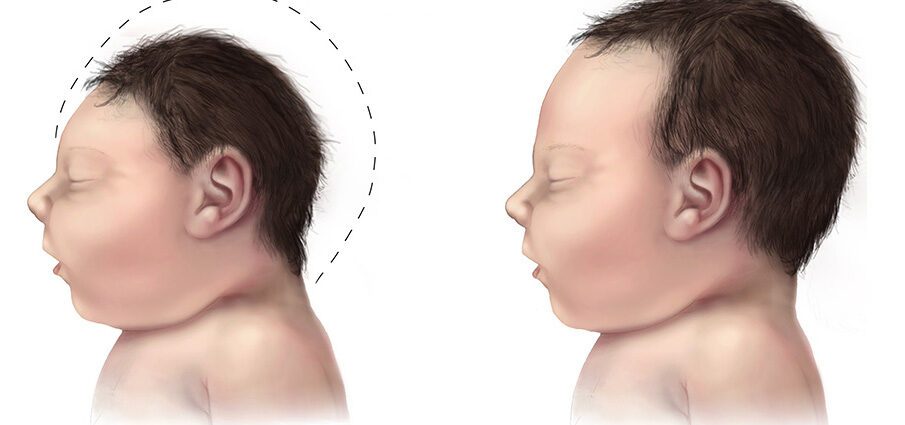
Mikrotsefaaliat iseloomustab kraniaalse ümbermõõdu areng sündides vähem kui tavaliselt. Mikrotsefaaliaga sündinud imikutel on tavaliselt väike aju suurus, mis seetõttu ei saa korralikult areneda. (1)
Haiguse levimus (juhtumite arv antud populatsioonis teatud ajahetkel) on siiani teadmata. Lisaks on näidatud, et haigus esineb sagedamini Aasias ja Lähis -Idas, esinemissagedus on 1/1 aastas. (000)
Mikrotsefaalia on seisund, mis on määratletud beebi pea suurusega, mis on tavalisest väiksem. Raseduse ajal kasvab lapse pea normaalselt tänu aju järkjärgulisele arengule. See haigus võib seejärel areneda raseduse ajal, lapse aju ebanormaalse arengu ajal või sündides, kui selle areng järsult peatub. Mikrotsefaalia võib olla iseenesest tagajärg, ilma et lapsel oleks muid kõrvalekaldeid või muul viisil seotud muude sündimisel nähtavate puudustega. (1)
On haiguse raske vorm. See tõsine vorm ilmneb aju ebanormaalse arengu tagajärjel raseduse ajal või sündides.
Mikrotsefaalia võib seega esineda lapse sündimisel või areneda esimestel kuudel pärast sünnitust. See haigus on sageli tingitud geneetilistest kõrvalekalletest, mis häirivad ajukoore kasvu loote arengu esimestel kuudel. See patoloogia võib olla ka uimastite või alkoholi kuritarvitamise tagajärg emal raseduse ajal. Haiguse allikaks võivad olla ka ema nakatumised tsütomegaloviiruse, punetiste, tuulerõugete jt.
Ema Zika viirusega nakatumise korral on viiruse levik nähtav ka lapse kudedes, mis viib ajusurmani. Selles kontekstis seostatakse neerukahjustusi sageli Zika viirusega.
Haiguse tagajärjed sõltuvad selle tõsidusest. Tõepoolest, lastel, kellel areneb mikrotsefaalia, võivad esineda kognitiivse arengu kahjustused, motoorsete funktsioonide hilinemine, keelelised raskused, lühike kehaehitus, hüperaktiivsus, epilepsiahood, koordinatsioonihäired või isegi muud neuroloogilised kõrvalekalded. (2)
Sümptomid
Mikrotsefaaliat iseloomustab pea suurus, mis on tavalisest väiksem. See anomaalia on aju arengu vähenemise tagajärg looteperioodil või pärast sünnitust.
Mikrotsefaaliaga sündinud imikutel võib olla mitmeid kliinilisi ilminguid. Need sõltuvad otseselt haiguse tõsidusest ja hõlmavad järgmist: (1)
- epileptilised krambid;
- lapse vaimse arengu, rääkimise, kõndimise jms viivitused;
- vaimupuue (vähenenud õppimisvõime ja elutähtsate funktsioonide hilinemine);
- koordinatsioonihäired;
- neelamisraskused;
- kuulmislangus;
- silmaprobleemid.
Need erinevad sümptomid võivad kogu subjekti elu jooksul ulatuda kergest kuni raskeni.
Haiguse päritolu
Mikrotsefaalia on tavaliselt lapse aju hilinenud arengu tagajärg, mistõttu pea ümbermõõt on tavalisest väiksem. Sellest seisukohast, kus aju areng on raseduse ja lapsepõlve ajal tõhus, võib nende kahe eluperioodi jooksul tekkida mikrotsefaalia.
Teadlased on välja pakkunud haiguse erinevaid põhjuseid. Nende hulka kuuluvad teatud nakkused raseduse ajal, geneetilised kõrvalekalded või isegi alatoitumine.
Lisaks on mikrotsefaalia tekkega seotud ka mõned järgmistest geneetilistest haigustest:
- Cornelia de Lange sündroom;
- kassi sündroomi nutmine;
- Downi sündroom;
- Rubinsteini - Taybi sündroom;
- Seckeli sündroom;
-Smith-Lemli-Opitzi sündroom;
- trisoomia 18;
- Downi sündroom.
Muud haiguse põhjused on järgmised: (3)
- kontrollimatu fenüülketonuuria (PKU) emal (fenüülalaniinhüdroksülaasi (PAH) ebanormaalsuse tagajärg, fenüülalaniini tootmise suurenemine plasmas ja toksiline toime ajule);
- metüülelavhõbeda mürgistus;
- kaasasündinud punetised;
- kaasasündinud toksoplasmoos;
- kaasasündinud tsütomegaloviiruse (CMV) infektsioon;
- teatud ravimite, eriti alkoholi ja fenütoiini kasutamine raseduse ajal.
Samuti on tõestatud, et laste Zica viirusega nakatumine lastel põhjustab mikrotsefaalia arengut. (1)
Riskifaktorid
Mikrotsefaaliaga seotud riskitegurid hõlmavad seega ema nakkuste kogumit, pärilikke või mitte pärilikke geneetilisi kõrvalekaldeid, kontrollimatu fenüülketonuuria emal, kokkupuude teatud kemikaalidega (nt metüülelavhõbe) jne.
Ennetamine ja ravi
Mikrotsefaalia saab diagnoosida raseduse ajal või vahetult pärast lapse sündi.
Raseduse ajal saab ultraheliuuringu abil kindlaks teha haiguse võimaliku esinemise. See test viiakse tavaliselt läbi raseduse teisel trimestril või isegi kolmanda trimestri alguses.
Pärast lapse sündi mõõdavad meditsiiniseadmed beebi pea ümbermõõdu keskmist suurust (peaümbermõõt). Saadud mõõtmist võrreldakse seejärel elanikkonna keskmistega vanuse ja soo funktsioonina. See sünnijärgne test tehakse tavaliselt vähemalt 24 tundi pärast sünnitust. See periood võimaldab tagada sünnituse ajal kokkusurutud kolju õige moodustumise.
Kui kahtlustatakse mikrotsefaalia esinemist, on diagnoosi kinnitamiseks või mitte kinnitamiseks võimalik teha muid täiendavaid uuringuid. Nende hulka kuuluvad eelkõige skanner, MRI (magnetresonantstomograafia) jne.
Haiguse ravi kestab kogu patsiendi elu. Praegu ei ole välja töötatud ühtegi ravivat ravimit.
Kuna haiguse raskusaste on erinevatel lastel erinev, ei ole healoomulisel kujul imikutel muid sümptomeid kui pea ümbermõõt. Seetõttu jälgitakse neid haigusjuhtumeid tähelepanelikult ainult lapse arengu ajal.
Haiguse raskemate vormide korral vajavad lapsed seekord ravi, mis võimaldab võidelda perifeersete probleemidega. Nende laste intellektuaalsete ja füüsiliste võimete parandamiseks ja maksimeerimiseks on olemas terapeutilised vahendid. Samuti võib välja kirjutada ravimeid krampide ja muude kliiniliste ilmingute vältimiseks. (1)
Haiguse prognoos on üldiselt hea, kuid sõltub suuresti haiguse tõsidusest. (4)