









Sisu
Bourneville'i tuberoosne skleroos
Mis see on?
Bourneville'i mugulskleroos on keeruline geneetiline haigus, mida iseloomustab healoomulise (mittevähkkasvaja) kasvaja tekkimine erinevates kehaosades. Need kasvajad võivad seejärel paikneda nahas, ajus, neerudes ja muudes elundites ja kudedes. See patoloogia võib põhjustada ka tõsiseid probleeme inimese arengus. Siiski on haiguse kliinilised ilmingud ja raskusaste patsienditi erinev.
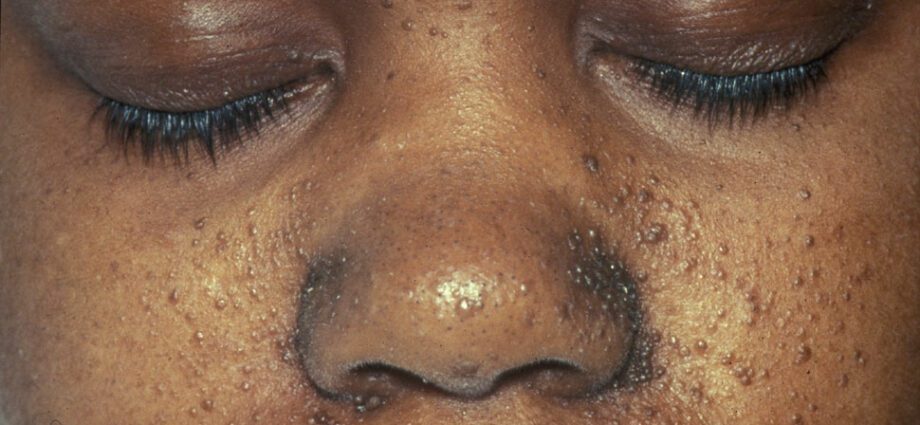
Seotud nahaanomaaliad on üldiselt sarnased täppidega nahal või piirkondadega, kus nahk on heledam kui ülejäänud kehal. Kasvajate teket näol nimetatakse angiofibroomiks.
Ajukahjustuse kontekstis on kliinilisteks tunnusteks epilepsiahood, käitumisprobleemid (hüperaktiivsus, agressiivsus, intellektihäired, õpiraskused jne). Mõnedel seda haigust põdevatel lastel on isegi teatud vormis autism, arenguhäired, mis mõjutavad sotsiaalset suhtlust ja suhtlemist. Healoomulised ajukasvajad võivad samuti põhjustada tüsistusi, mis võivad katsealusele surmaga lõppeda.
Tuumorite tekkimine neerudes on tuberoosskleroosiga inimestel tavaline. See võib põhjustada tõsiseid neerufunktsiooni tüsistusi. Lisaks võivad kasvajad areneda südames, kopsudes ja võrkkestas. (2)
See on haruldane haigus, mille levimus (juhtude arv antud populatsioonis teatud ajahetkel) ulatub 1/8 kuni 000/1 inimeseni. (15)
Sümptomid
Bourneville'i tuberoosskleroosiga seotud kliinilised ilmingud varieeruvad olenevalt mõjutatud organitest. Lisaks on haigusega seotud sümptomid inimestel väga erinevad. Sümptomid ulatuvad kergest kuni raskeni.
Selle haiguse kõige levinumad sümptomid on epilepsiahood, kognitiivsed ja käitumishäired, nahaanomaaliad jne. Kõige sagedamini kannatavad organid: aju, süda, neerud, kopsud ja nahk.
Selle haiguse korral on võimalik pahaloomuliste (vähi) kasvajate teke, kuid need on haruldased ja mõjutavad peamiselt neere.
Haiguse kliinilised tunnused ajus tulenevad erinevatel tasanditel esinevatest rünnakutest:
- kortikaalsete tuberkulooside kahjustus;
– ependümaalsed sõlmed (SEN);
- hiiglaslikud ependümaalsed astrotsütoomid.
Nende tagajärjeks on: vaimne alaareng, õpiraskused, käitumishäired, agressiivsus, tähelepanuhäired, hüperaktiivsus, obsessiiv-kompulsiivsed häired jne.
Neerukahjustust iseloomustab tsüstide või angiomüolipoomide areng. Need võivad põhjustada neeruvalu ja isegi neerupuudulikkust. Kui raske verejooks on märgatav, võib selle põhjuseks olla raske aneemia või kõrge vererõhk. Nähtavad võivad olla ka muud tõsisemad, kuid haruldased tagajärjed, eelkõige kartsinoomide (epiteeli moodustavate rakkude kasvaja) teke.
Silmakahjustus võib sarnaneda võrkkesta nähtavate laikudega, põhjustades nägemishäireid või isegi pimedaksjäämist.
Naha kõrvalekaldeid on palju:
– hüpomelanilised laigud: mille tulemuseks on heledate laikude ilmumine nahale kõikjal kehal, mis on tingitud nahale värvi andva valgu melaniini puudusest;
- punaste laikude ilmumine näole;
- värvunud laigud otsmikul;
- muud ühelt inimeselt sõltuvad nahaanomaaliad.
Kopsukahjustused esinevad 1/3 patsientidest, kus naised on vähesel määral ülekaalus. Kaasnevad sümptomid on siis enam-vähem tõsised hingamisraskused.
Haiguse päritolu
Haiguse päritolu on geneetiline ja pärilik.
Ülekanne hõlmab mutatsioone TSC1 ja TSC2 geenides. Need huvipakkuvad geenid tulevad mängu valkude moodustumisel: hamartiin ja tuberiin. Need kaks valku võimaldavad interaktiivse mängu kaudu rakkude proliferatsiooni reguleerida.
Seda haigust põdevatel patsientidel on igas rakus vähemalt üks nende geenide muteerunud koopia. Need mutatsioonid piiravad seejärel hamartiini või tuberkuliini moodustumist.
Kontekstis, kus geeni kaks koopiat on muteerunud, takistavad need täielikult nende kahe valgu tootmist. Seetõttu ei võimalda see valgupuudus organismil enam teatud rakkude kasvu reguleerida ja põhjustab selles mõttes kasvajarakkude arengut erinevates kudedes ja/või elundites.
Riskifaktorid
Sellise patoloogia tekke riskitegurid on geneetilised.
Tõepoolest, haiguse edasikandumine on efektiivne autosomaalse domineeriva režiimi kaudu. Mõlemal juhul asub huvipakkuv muteerunud geen mitteseksuaalses kromosoomis. Lisaks piisab haiguse arenemiseks ainult ühe muteerunud geeni kahest koopiast.
Selles mõttes on indiviidil, kellel on üks kahest selle haiguse all kannatavast vanemast, 50% risk haige fenotüübi tekkeks ise.
Ennetamine ja ravi
Haiguse diagnoos on ennekõike diferentseeritud. See põhineb ebatüüpilistel füüsilistel kriteeriumidel. Enamasti on haiguse esimesed iseloomulikud tunnused: korduvate epilepsiahoogude esinemine ja katsealuse arengu hilinemine. Muudel juhtudel põhjustavad need esimesed märgid nahalaike või südamekasvaja tuvastamist.
Pärast esimest diagnoosi on diagnoosi kinnitamiseks või mitte kinnitamiseks hädavajalikud täiendavad uuringud. Need sisaldavad:
- aju skaneerimine;
- aju MRI (magnetresonantstomograafia);
- südame, maksa ja neerude ultraheliuuring.
Diagnoos võib olla tõhus lapse sündimisel. Vastasel juhul on oluline, et see toimuks võimalikult kiiresti, et patsient võimalikult kiiresti enda kätte võtta.
Praegu ei ole haiguse vastu ravi. Seotud ravimeetodid ei sõltu seetõttu iga inimese sümptomitest.
Tavaliselt antakse krambihoogude piiramiseks epilepsiavastaseid ravimeid. Lisaks on ette nähtud ka ravimid aju ja neerude kasvajarakkude raviks. Käitumisprobleemide kontekstis on vajalik lapse spetsiifiline kohtlemine.
Haiguse ravi on tavaliselt pikaajaline. (1)